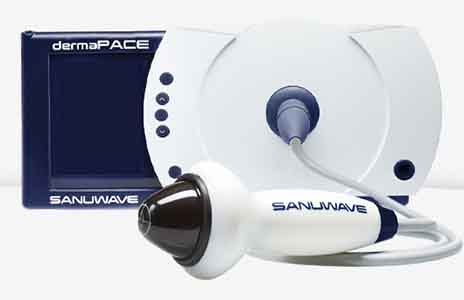
Dermapace System, the first shock wave device intended to treat diabetic foot ulcers.
“Diabetes is the leading cause of lower limb amputations,” said Binita Ashar, M.D., director of the division of surgical devices in FDA’s Center for Devices and Radiological Health. “The FDA is dedicated to making technologies available that can help improve the quality of life for those with chronic diseases. Additional options for successfully treating and healing ulcer wounds may help prevent lower limb amputations.”
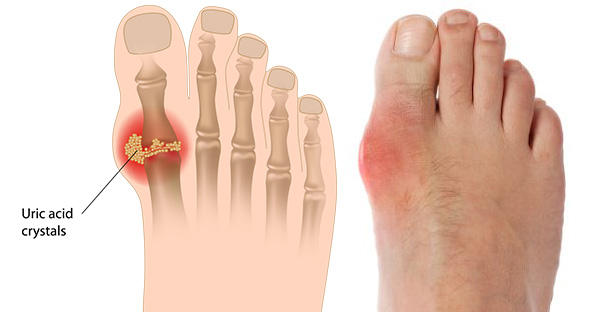
An estimated 30.3 million people in the United States have been diagnosed with diabetes, according to the Centers for Disease Control and Prevention. Diabetes damages blood vessels and nerves, particularly in the feet, and can lead to severe infections that are difficult to treat. About 25 percent of people with diabetes will experience a foot ulcer in their lifetime. Amputation is sometimes necessary when circulation is so poor that a foot ulcer fails to heal or when treatment fails to stop the spread of an infection.
The Dermapace System is intended to be used in the treatment of chronic, full-thickness diabetic foot ulcers with wound areas measuring no larger than 16 cm2 (about the size of a soda can top) which extend through the epidermis, dermis, tendon, or capsule, but without bone exposure. The Dermapace System is an external (extracorporeal) shock wave system that uses pulses of energy, similar to sound waves, to mechanically stimulate the wound. The device is intended for adult patients (22 years and older), presenting with diabetic foot ulcers lasting for more than 30 days, and should be used along with standard diabetic ulcer care.
The FDA reviewed clinical data from two multi-center, randomized, double-blind studies with a total of 336 diabetic patients receiving either usual care, which includes wet-to-dry dressings or debridement (removal of damaged tissue) as needed, plus the Dermapace System shock wave therapy or usual care plus non-working (sham) shock wave therapy. Both patient groups included those with poorly controlled and well-controlled blood glucose levels.
The patients who had between one and seven treatments with the Dermapace System showed an increase in wound healing at 24 weeks with a 44 percent wound closure rate. Those patients treated with the sham shock wave therapy showed a 30 percent wound closure rate during the same time period.
The most common side effects observed were pain during application of the device, local bruising and numbness, migraines, nausea, fainting, wound infection, infection beyond the wound (cellulitis, osteomyelitis) and fever.