This course provides a general introduction to COVID-19 and
emerging respiratory viruses and is intended for public health professionals,
incident managers and personnel working for the United Nations, international
organizations and NGOs.
All you need to sign up is a valid email address and you can
learn from anywhere in the world, at any time
About OpenWHO
OpenWHO is WHO’s new interactive, web-based,
knowledge-transfer platform offering online courses to improve the response to
health emergencies.
For details visit OpenWHO.org
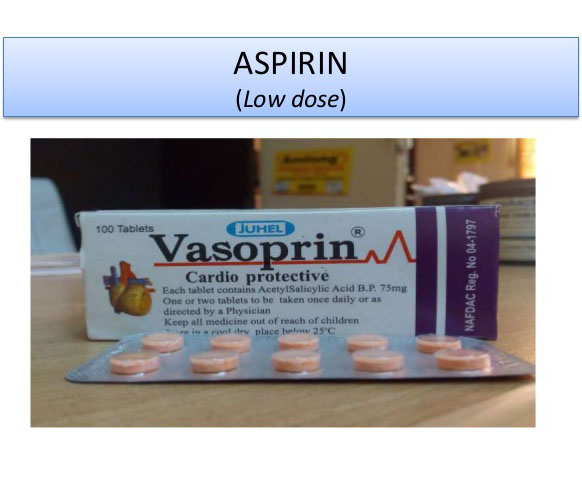
Babies delivered
before the 37th week of pregnancy have higher rates of infant mortality. This
is especially true for infants born in low- and middle-income countries (LMICs).
Prior studies have found that pregnant women who receive low-dose aspirin daily
are less likely to deliver their babies prematurely, particularly when the
medication is started before the fourth month of pregnancy.
An article recently published in The Lancet reviewed the results
of a randomised, double-masked, placebo-controlled trial. The goal of the study was to determine whether
81 mg of aspirin reduces the risk of premature delivery in women with no
previous births (i.e. nulliparous). Between March 23, 2016
and April 11, 2019, approximately 12,000 nulliparous pregnant women in six LMICs
received daily doses of either 81 mg of aspirin or placebo. Enrolled
participants were early in their pregnancy (at least 6 weeks) and received the
study medication until either delivery or the ninth month (37th
week) of pregnancy.
Low-dose Aspirin Reduced
the Risk of Preterm Birth
The results of the study showed that when compared to placebo, low-dose aspirin reduced the rates of preterm delivery (13.1% vs 11.6%; p=0.012), perinatal mortality (53.6% vs 45.7%; p=0.048), and foetal death (between the fourth month of pregnancy and 7 days postpartum: 60.8% vs 52.1%; p=0.039). Both treatment groups had similar rates of low birth-weight and other adverse effects. Some of these adverse effects include bleeding (gastrointestinal and vaginal), haemorrhage (antepartum and postpartum), anaemia, congenital anomaly, and maternal death.
The authors noted that
an optimal dose of aspirin for this patient population needs to be determined,
as higher doses may provide additional benefits. Also noted is the fact that
this is the first large randomised-controlled trial to investigate this theory
and the results confirm the findings from previous studies.
In summary, these
results suggest that aspirin should be considered as a treatment option to
prevent preterm birth in pregnant women.
References:
Hoffman MK, et al. Low-dose aspirin for the prevention of preterm delivery in nulliparous women with a singleton pregnancy (ASPIRIN): a randomised, double-blind, placebo-controlled trial. The Lancet, 395(10220), pp.285-293. doi: 10.1016/S0140-6736(19)32973-3
[FDA Drug Safety Communication, January 28, 2020] – The U.S.
FDA strengthens warning that constipation caused by clozapine can, uncommonly,
progress to serious bowel complications. This can lead to hospitalization or
even death if constipation is not diagnosed and treated quickly.
Clozapine affects how the intestines function in the
majority of patients. It produces effects ranging from constipation, which is a
common occurrence, to serious but uncommon bowel problems, including complete
blockage of the bowel.
FDA recommends that health care professionals should:
Evaluate bowel function before starting a patient on clozapine.
Avoid co-prescribing clozapine with other anticholinergic medicines that can cause gastrointestinal hypomotility.
Advise patients frequently of the significant risk of constipation and life-threatening bowel issues and the need to stay hydrated to prevent constipation.
Question patients about the frequency and quality of their bowel movements throughout treatment.
Advise patients to contact a health care professional right away if they have difficulty having a bowel movement or passing stools, do not have a bowel movement at least three times a week or less than their normal frequency, or are unable to pass gas.
Monitor patients for symptoms of potential complications associated with gastrointestinal hypomotility such as nausea, abdominal distension or pain, and vomiting.
Consider prophylactic laxative treatment when starting clozapine in patients with a history of constipation or bowel obstruction.
Clozapine can cause severe neutropenia, which can lead to
serious and fatal infections. Its use should be limited to patients:
with schizophrenia who are non-responsive to, or intolerant of, classical antipsychotic agents, or with schizophrenia or schizoaffective disorder who are at risk of recurrent suicidal behaviour,
who have initially normal leukocyte findings (white blood cell count (WBC) ≥3500/mm³ (3.5 x 109/L), and absolute neutrophil counts (ANC) ≥2000/mm³ (2.0 x 109/L)),
and in whom regular white blood cell counts and absolute neutrophil counts can be performed as follows: weekly during the first 18 weeks of therapy, and at least every 4 weeks thereafter throughout treatment. Monitoring must continue throughout treatment and for 4 weeks after complete discontinuation of Clozapine.
Patients must be advised to immediately report symptoms
consistent with severe neutropenia or infection (e.g., fever, weakness,
lethargy, or sore throat).
2. Increased mortality in elderly patients with dementia-related psychosis
Elderly patients with dementia-related psychosis treated
with antipsychotic drugs are at an increased risk of death. Clozapine is not
approved for this condition.
3. Orthostatic hypotension, bradycardia, syncope
Clozapine has alpha-blocking activity and can cause
orthostatic hypotension, with or without syncope. Rarely, collapse can be
profound and may be accompanied by cardiac and/or respiratory arrest. The risk
is dose-related and highest during the initial titration period, particularly
with rapid dose escalation. These reactions can occur with the first dose, with
doses as low as 12.5 mg per day. Initiate treatment at 12.5 mg once or twice
daily; titrate slowly; and use divided dosages. Use Clozapine cautiously in
patients with cardiovascular or cerebrovascular disease or conditions
predisposing to hypotension (e.g., dehydration, use of antihypertensive
medications).
4. Seizures
Clozapine may lower seizure threshold. The risk is
dose-related. Initiate treatment at 12.5 mg, titrate gradually, and use divided
dosing. Use caution when administering Clozapine to patients with a history of
seizures or other predisposing risk factors for seizure (CNS pathology,
medications that lower the seizure threshold, alcohol abuse). Caution patients
about engaging in any activity where sudden loss of consciousness could cause
serious risk to themselves or others.
5. Myocarditis, cardiomyopathy and mitral valve incompetence
Fatal myocarditis and cardiomyopathy have occurred with Clozapine
treatment. Discontinue Clozapine and obtain cardiac evaluation if findings
suggest these cardiac reactions. Tachycardia that persists at rest, accompanied
by arrhythmias, shortness of breath or signs and symptoms of heart failure, may
rarely occur during the first month of treatment and very rarely thereafter.
The occurrence of these signs and symptoms necessitates an urgent diagnostic
evaluation for myocarditis, especially during the titration period. Consider
the possibility of myocarditis in patients receiving Clozapine who present with
unexplained fatigue, dyspnoea, tachypnoea, fever, chest pain, tachycardia,
palpitations, other signs and symptoms of heart failure, ECG changes (such as
ST-T wave abnormalities) or arrhythmias.
Generally, patients with Clozapine-related myocarditis or cardiomyopathy should not be rechallenged with Clozapine
[FDA News Release, January 31, 2020] – The U.S. Food and
Drug Administration (FDA) has approved Palforzia [Peanut (Arachis hypogaea)
Allergen Powder-dnfp] to mitigate allergic reactions, including anaphylaxis,
that may occur with accidental exposure to peanuts. Treatment with Palforzia
may be initiated in individuals ages 4 through 17 years with a confirmed
diagnosis of peanut allergy and may be continued in individuals 4 years of age
and older. Those who take Palforzia must continue to avoid peanuts in their
diets.
Peanut allergy has no cure, allergic individuals
must strictly avoid exposure to prevent severe and potentially life-threatening
reactions.
Even with strict avoidance, inadvertent
exposures can and do occur.
Palforzia, in combination with peanut avoidance,
provides an FDA-approved treatment option to help reduce the risk of these
allergic reactions in children with peanut allergy.
Palforzia cannot be used for the emergency
treatment of allergic reactions, including anaphylaxis.
Initial Dose Escalation, and the first dose of
each Up-Dosing level, are administered under supervision of a healthcare
professional in a healthcare setting with the ability to manage potentially
severe allergic reactions, including anaphylaxis.
Palforzia should not be administered to those
with uncontrolled asthma.
Patients or their parents or caregivers must
also be counseled on the need for the patients to have injectable epinephrine
available for immediate use at all times, the need for continued dietary peanut
avoidance, and how to recognize the signs and symptoms of anaphylaxis
The FDA granted approval of Palforzia to Aimmune
Therapeutics.
Peanut allergy is a condition in which the body’s immune
system mistakenly identifies even small amounts of peanut as harmful. Allergic
reactions to peanut are unpredictable in occurrence and in how they present,
with some individuals experiencing severe reactions from even trace amounts.
Physical symptoms can develop within seconds of exposure and may include skin
reactions (e.g., hives, redness or swelling), digestive discomfort, or more
dangerous reactions, such as constriction of the throat and airways, and loss
of adequate blood flow to vital organs of the body. Antihistamines and
epinephrine can be used to treat allergic reactions, but severe reactions can
be fatal even with appropriate, prompt treatment. Palforzia cannot be used
for the emergency treatment of allergic reactions, including anaphylaxis.
Treatment with Palforzia consists of three phases: Initial
Dose Escalation, Up-Dosing, and Maintenance. The Initial Dose Escalation phase
is given on a single day. The Up-Dosing phase consists of 11 increasing dose
levels and occurs over several months. Initial Dose Escalation, and the
first dose of each Up-Dosing level, are administered under supervision of a
healthcare professional in a healthcare setting with the ability to manage
potentially severe allergic reactions, including anaphylaxis. While
anaphylaxis can occur at any time during Palforzia therapy, patients are at
highest risk during and after the Initial Dose Escalation and the first dose of
each Up-Dosing level. During Up-Dosing, if the patient tolerates the first dose
of an increased dose level, the patient may continue that dose level daily at
home. After a patient completes all Up-Dosing levels, they may begin the daily
maintenance dose. Patients who experience certain allergic reactions due to
Palforzia may need to discontinue treatment or have their dosing schedule
modified.
Palforzia is a powder that is manufactured from peanuts and
packaged in pull-apart color-coded capsules for Dose Escalation and Up-Dosing,
and in a sachet for maintenance treatment. The powder is emptied from the
capsules or sachet and mixed with a small amount of semisolid food – such as
applesauce, yogurt, or pudding – that the patient then consumes.
The effectiveness of Palforzia is supported by a randomized,
double-blind, placebo-controlled study conducted in the U.S., Canada and Europe
in approximately 500 peanut-allergic individuals. Effectiveness was assessed by
evaluating the percentage of study participants tolerating an oral challenge
with a single 600 mg dose of peanut protein (twice the daily maintenance dose
of Palforzia) with no more than mild allergic symptoms after 6 months of
maintenance treatment. The results showed that 67.2% of Palforzia recipients
tolerated a 600 mg dose of peanut protein in the challenge, compared to 4.0% of
placebo recipients.
The safety of Palforzia was assessed in two double-blind,
placebo-controlled studies in approximately 700 peanut-allergic individuals.
The most commonly reported side effects of Palforzia were abdominal pain,
vomiting, nausea, tingling in the mouth, itching (including in the mouth and
ears), cough, runny nose, throat irritation and tightness, hives, wheezing and
shortness of breath and anaphylaxis. Palforzia should not be administered to
those with uncontrolled asthma.
To mitigate the risk of anaphylaxis associated with Palforzia, the FDA is requiring a Risk Evaluation and Mitigation Strategy (REMS) with this approval, which includes elements to assure safe use. Palforzia will only be available through specially certified healthcare providers, health care settings, and pharmacies to patients who are enrolled in the REMS program. The FDA is requiring that healthcare providers who prescribe Palforzia – and healthcare settings that dispense and administer Palforzia – are educated on the risk of anaphylaxis associated with its use. In addition, the Initial Dose Escalation phase and first dose of each Up-Dosing level must only be administered to patients in a certified healthcare setting equipped to monitor patients and to identify and manage anaphylaxis. Patients or their parents or caregivers must also be counseled on the need for the patients to have injectable epinephrine available for immediate use at all times, the need for continued dietary peanut avoidance, and how to recognize the signs and symptoms of anaphylaxis.
[NAFDAC Public Alert No: 0023/2019, December 29, 2019] – Alert
on Dangerous and Unapproved Use of Paracetamol Tablets in Food Preparation.
The National Agency for Food and Drug Administration and
Control (NAFDAC) hereby alerts the public on the dangerous and unapproved use
of Paracetamol Tablets to soften meat used in food preparation.
The members of the public, especially restaurant operators
are cautioned to desist from the dangerous and unapproved use of Paracetamol
Tablets to soften meat used in food preparation, as such illegal practice makes
food to be toxic, unwholesome and unfit for human consumption. When used to
cook, paracetamol is broken down (or hydrolyzed) into a toxic substance. This
substance ultimately damages the liver and some other organs in the body.
Thus, the consumption of toxic and unwholesome food illegally prepared using Paracetamol Tablets may result in serious health consequences, including liver damage, kidney failure and untimely death.
NAFDAC recommends the following safer alternative methods
for tenderizing meat:
Use genuine NAFDAC registered Table Salt (in
moderation) by soaking meat in salted water for about 30 minutes prior to cooking.
Cooking with a pressure cooker.
Marinating (soaking) with vinegar, citrus juices
or wine before cooking.
Marinating with enzymes e.g., Pineapple, kiwi,
ginger, Asian pear and pawpaw which contain enzymes that can help soften meat.
Slow cooking the meat, or
Using commercial meat tenderizers (in
moderation), available in powder or liquid form.
Genuine Paracetamol Tablets registered by NAFDAC are used to
relieve mild pain and if the pain persists, the patient should consult a
healthcare professional for expert advice.
NAFDAC is increasing surveillance on restaurant operators nationwide
and is urging members of the public to contact the nearest NAFDAC Office with
any information on dangerous and illegal use of Paracetamol Tablets in food
preparation.
Anybody or organization discovered to be involved in the
dangerous and illegal use of Paracetamol Tablets in food preparation will be
severely sanctioned.
Consumers are advised to report adverse events related to dangerous and illegal use of Paracetamol Tablets in food preparation to the nearest NAFDAC office, 0800-1-NAFDAC (0800-1-623322) TOLL FREE from all networks or via pharmacovigilance@nafdac.gov.ng
When used with CNS depressants or in patients with lung problems
[FDA Drug Safety Communication, December 19, 2019] – The
U.S. Food and Drug Administration (FDA) is warning that serious,
life-threatening, and fatal respiratory depression has been reported with the
gabapentinoids, gabapentin and pregabalin. Most cases occurred in association
with co-administered central nervous system (CNS) depressants, especially
opioids, in the setting of underlying respiratory impairment, or in the elderly.
Gabapentin and pregabalin are FDA-approved for a variety of
conditions, including seizures, nerve pain, and restless legs syndrome.
Our evaluation of respiratory depression with the
gabapentinoids provides some evidence contrary to the widely held belief that
gabapentinoids lack drug interactions and have wide therapeutic indices.
Published studies demonstrate these drugs can behave in an additive way to
potentiate central nervous system (CNS) and respiratory depression. CNS
depressants include opioids, anti-anxiety medicines, antidepressants, and
antihistamines. There is less evidence supporting the risk of serious breathing
difficulties in healthy individuals taking gabapentinoids alone.
What should patients and caregivers do?
Patients and caregivers should seek medical attention
immediately if you or someone you are caring for experiences symptoms of
respiratory problems, because these can be life-threatening. Symptoms to watch
for include:
Confusion or disorientation
Unusual dizziness or lightheadedness
Extreme sleepiness or lethargy
Slowed, shallow, or difficult breathing
Unresponsiveness, which means a person doesn’t
answer or react normally or you can’t wake them up
Bluish-colored or tinted skin, especially on the
lips, fingers, and toes
Always inform your health care professional about all the
drugs you are taking, including prescription and over-the-counter (OTC)
medicines and other substances such as alcohol.
What should health care professionals do?
When co-prescribing gabapentinoids with another CNS
depressant, particularly an opioid, or in patients with underlying respiratory
impairment, initiate the gabapentinoid at the lowest dose.
Adjust the dose of both gabapentin and pregabalin in
patients with renal impairment and patients undergoing hemodialysis, because
both drugs are excreted by the kidneys.
Monitor for symptoms of respiratory depression and sedation,
especially when co-prescribing gabapentinoids with an opioid or other CNS
depressant such as a benzodiazepine or when prescribing to patients with
underlying respiratory impairment, or elderly patients.
The management of respiratory depression may include close
observation, supportive measures, and reduction or withdrawal of CNS
depressants, including the gabapentinoid. Gabapentinoids used for analgesia or
seizure control should be tapered prior to discontinuation. See the prescribing
information for specific tapering guidance.
The gabapentinoid prescribing information already includes
guidance for health care professionals to caution patients about dizziness,
somnolence, and the potential for impaired ability to operate a car or complex
machinery.
The long-lasting capsule can remain in the stomach and release contraceptive drugs over several weeks.
December 4, 2019. By Anne Trafton | MIT News Office
Oral contraceptives are one of the most popular forms of
birth control: In the United States, about 12 percent of women between 15 and
49 use them. However, their effectiveness depends on being taken every day, and
it is estimated that about 9 percent of women taking birth control pills become
pregnant each year.
MIT researchers are now developing an oral contraceptive
that only has to be taken once a month, which could reduce unintended
pregnancies that result from forgetting to take a daily dose. This kind of
monthly contraceptive could have a significant impact on the health of women
and their families, especially in the developing world, the researchers say.
“We are hopeful that this work — the first example ever of a month-long pill or capsule to our knowledge — will someday lead to potentially new modalities and options for women’s health as well as other indications,” says Robert Langer, the David H. Koch Institute Professor at MIT.
The new contraceptive is contained within a gelatin-coated
capsule and can carry three weeks’ worth of a contraceptive drug. This capsule
remains in the stomach after being swallowed and gradually releases the drug.
Tests in pigs showed that this kind of drug release can achieve the same
concentration of the drug in the bloodstream as taking a daily dose.
Langer and Giovanni Traverso, an assistant professor of
mechanical engineering at MIT and a gastroenterologist at Brigham and Women’s
Hospital, are the senior authors of the study, which appears today in Science
Translational Medicine. Ameya Kirtane, a senior postdoc at MIT’s Koch Institute
for Integrative Cancer Research, and Tiffany Hua, a former technical associate
at MIT, are the lead authors of the paper.
Long-term delivery
The new contraceptive pill is based on star-shaped drug
delivery systems that the MIT team previously developed, which can remain in
the digestive tract for days or weeks after being swallowed. The delivery
systems are placed in gelatin capsules that dissolve once they reach the
stomach, allowing the folded arms of the star to expand and slowly release
their payload.
After being swallowed, the capsule unfolds and slowly releases its drug payload in the stomach. After a few weeks, it breaks down and moves through the digestive tract. Credits: Lyndra Therapeutics
In their earlier studies, the researchers loaded the
capsules with drugs to treat malaria, as well as HIV drugs, which currently
have to be taken every day. Much of this work has been funded by the Bill and
Melinda Gates Foundation, which urged the team to adapt the capsule to deliver
long-lasting contraceptive drugs. Previous research has suggested that people
are better at remembering to take medicine when they have to take it only
weekly or monthly, instead of daily.
To make their new contraceptive pill last for three to four
weeks, the researchers had to incorporate stronger materials than those used in
the earlier versions, which could survive in the harsh environment of the
stomach for up to two weeks. The researchers tested materials by soaking them
in simulated gastric fluid, which is very acidic, and found that two types of
polyurethane worked best for the arms and the central core of the star.
The researchers loaded the contraceptive drug levonorgestrel
into the arms of the device and found that by changing the concentrations of
the polymers that they mix with the drug, they can control the rate at which it
is released. Once the capsule reaches the stomach it expands and becomes lodged
in place.
In a study of pigs, the researchers found that the capsules
could release the drug at a fairly constant rate for up to four weeks. The
concentration of the drug found in the pigs’ bloodstream was similar to the
amount that would be present after ingesting daily levonorgestrel tablets.
However, the capsules maintained these drug levels for nearly a month, while
the tablets last for only a day.
For use in humans, the capsule would be designed to break
down after three or four weeks and exit the body through the digestive tract.
The researchers are working on several possible ways to trigger the arms to
break off, including through changes in pH, changes in temperature, or exposure
to certain chemicals.
“Lack of access to contraceptives is a global health issue that contributes to unnecessary maternal and newborn deaths every year,” says Kimberly Scarsi, an associate professor of pharmacy practice and science at the University of Nebraska Medical Center, who was not involved in the research. “A once-monthly oral contraceptive would provide a discreet, noninvasive birth control option that could significantly improve medication adherence to give women more control over their health and family planning decisions.”
Health impact
Lyndra Therapeutics, a company founded by Langer, Traverso,
and others, recently received a $13 million grant from the Gates Foundation to
further develop the monthly contraceptive pill so that it can be tested in
humans.
“Through the development of these technologies, we aim to transform people’s experience with taking medications by making it easier, with more infrequent dosing in the first once-a-month, orally delivered drug system. We’re very committed to getting these technologies to people over the coming years,” says Traverso, who said he anticipates human tests may be possible within three to five years.
Improved contraception not only has health benefits, but
also makes it easier for women to go to school and financially support
themselves and their families. However, according to the World Health
Organization, 214 million women of reproductive age in developing countries who
want to avoid pregnancy are not using a modern contraceptive method, such as
birth control pills.
“Coming up with a monthly version of a contraceptive drug could have a tremendous impact on global health,” Kirtane says. “The impact that oral contraceptives can have on human health and gender equality cannot be overstated.”
The researchers also believe that such a pill could be
appealing for women who would prefer a long-lasting oral contraceptive over
other long-term contraceptives such as intrauterine devices.
“Even with all these long-acting devices available, there’s a certain population who prefers to take medications orally rather than have something implanted,” Kirtane says. “For those patients, something like this would be extremely helpful.”
The research was funded by the Bill and Melinda Gates
Foundation. Other MIT authors of the study are Alison Hayward, Aniket Wahane,
Aaron Lopes, Taylor Bensel, Sierra Brooks, Declan Gwynne, Jacob Wainer, Joy
Collins, and Siid Tamang. Ambika Bajpayee of Northeastern University and Frank
Stanczyk and Lihong Ma of the University of Southern California are also
authors of the paper.
The National Agency for Food and Drug Administration and
Control (NAFDAC) has issued public alerts and safety communication affecting
the following drug products in the months of November and December 2019.
[NAFDAC Public Alert No: 0022/2019, December 4, 2019] – Alert on Recall of Glosunate® Plus 50 (Artesunate 50mg and Amodiaquine 153mg) Tablets Batch Number GK18010
Reason for the recall: Unsatisfactory report of
laboratory analysis. Failed assay, dissolution and related substances tests.
Company Name: Uche St. Company Limited
Manufacturer: Globela Pharma Pvt, Gujarat, India
NAFDAC has taken action to prevent or stop the distribution
and sale of Glosunate Plus 50 tablets Batch Number GK18010 in Nigeria. The
Agency has also directed all distributors, wholesalers and retailers to submit
Glosunate Plus 50 tablets Batch Number GK18010 in their possession to the
nearest NAFDAC office.
[NAFDAC Public Alert No: 0021/2019, December 4, 2019] – Alert on Recall of Solartep® (Dihydroartemisinin 40mg/Piperaquine Phosphate 320mg) Tablets Batch Number F17S0826
Reason for the recall: Unsatisfactory report of
laboratory analysis. Failed assay and dissolutions tests.
Company Name: Solutions Pharmaceutical Nigeria
Limited
Manufacturer: Mingshen Pharmaceutical Factory, China
NAFDAC has taken action to prevent or stop the distribution
and sale of Solartep tablets Batch number F17S0826. The Agency has also directed
all distributors, wholesalers and retailers are to submit Solartep tablets
Batch number F17S0826 in their possession to the nearest NAFDAC office.
[NAFDAC Public Alert No: 0019/2019, November 3, 2019] – Alert on Voluntary Recall of Xalatan® (Latanoprost 0.005%) Eye Drops Lot Numbers W67369 and AK4753
Reason for the recall: Voluntary recall due to detection
of confirmed falsified Xalatan® 0.005% w/v Eye Drops 2.5 ml solution with
authentic Pfizer Specialties Limited Lot Numbers W67369 and AK4753. The
falsified Xalatan® 0.005% w/v Eye Drops 2.5 ml solution was detected following
a market survey conducted by Pfizer Global Security on selected Pfizer products
exposed to counterfeiting in the Nigerian market.
NAFDAC has taken steps to prevent the distribution and sale
of the falsified Xalatan® 0.005% w/v Eye Drops in Nigeria. The Agency has also
directed distributors and wholesalers in possession of Xalatan* (Latanoprost
0.005%) Eye Drops Lot Numbers W67369 and AK4753 to immediately stop their
distribution and sale.
[FDA News Release, November 25, 2019] – The U.S. Food and
Drug Administration (FDA) has granted accelerated approval for Oxbryta™ (voxelotor)
tablets for the treatment of sickle cell disease (SCD) in adults and children
12 years of age and older. Oxbryta, an oral therapy taken once daily, is the
first approved treatment that directly inhibits sickle hemoglobin
polymerization, the root cause of SCD.
Global Blood Therapeutics, Inc. (GBT), makers of Oxbryta™, today
announced that the medicine is expected to be available through its specialty
pharmacy partner network within two weeks.
“Today is a major milestone not only for GBT but, most
importantly, for people living with SCD, their families and those who care for
them. When we started our journey with the SCD community more than eight years
ago, we set out to transform the way this devastating, lifelong disease is
treated,” said Ted W. Love, M.D., president and chief executive officer of GBT.
“We are proud to bring this breakthrough therapy to the SCD community. Uniquely
developed from inception to treat SCD, Oxbryta embodies GBT’s commitment to
develop and deliver innovative medicines for patients with overlooked,
life-limiting chronic diseases. We are grateful to the patients, caregivers,
clinical trial investigators, healthcare providers and advocates who have
worked alongside us to develop this first-in-class therapy.”
SCD affects an estimated 100,000 people in the United States
and millions of people throughout the world, particularly among those whose
ancestors are from sub-Saharan Africa. It also affects people of Hispanic, South
Asian, Southern European and Middle Eastern ancestry. SCD is a lifelong
inherited blood disorder that impacts hemoglobin, a protein carried by red
blood cells that delivers oxygen to tissues and organs throughout the body. Due
to a genetic mutation, people with SCD form abnormal hemoglobin known as sickle
hemoglobin. Through a process called hemoglobin polymerization, red blood cells
become sickled – deoxygenated, crescent-shaped and rigid. The sickling process
causes hemolytic anemia (low hemoglobin due to red blood cell destruction) and
blockages in capillaries and small blood vessels, which impede the flow of
blood and oxygen throughout the body. The diminished oxygen delivery to tissues
and organs can lead to life-threatening complications, including stroke and
irreversible organ damage.
The accelerated approval of Oxbryta is based on clinically
meaningful and statistically significant improvements in hemoglobin levels,
accompanied by reductions in red blood cell destruction (hemolysis). Data from
the Phase 3 HOPE (Hemoglobin Oxygen Affinity Modulation to Inhibit HbS
PolymErization) Study of 274 patients 12 years of age and older with SCD showed
that, after 24 weeks of treatment, 51.1% of patients receiving Oxbryta achieved
a greater than 1 g/dL increase in hemoglobin compared with 6.5% receiving
placebo (p<0.001). Results from the HOPE Study were published in June 2019
in The New England Journal of Medicine.
The most common adverse reactions occurring in ≥10% of
patients treated with Oxbryta with a difference of >3% compared to placebo
were headache (26% vs. 22%), diarrhea (20% vs. 10%), abdominal pain (19% vs.
13%), nausea (17% vs. 10%), fatigue (14% vs. 10%), rash (14% vs. 10%) and
pyrexia (12% vs. 7%).
“SCD is a devastating, lifelong, inherited blood disorder
that greatly impacts a person’s life, including their ability to work, attend
school and look after their families, and it can reduce their overall life
expectancy,” said Beverley Francis-Gibson, president and CEO of the Sickle Cell
Disease Association of America. “After decades of waiting, we now have a
treatment option that could change the course of this disease. We look forward
to continuing to collaborate with GBT on initiatives aimed at transforming the
care of patients living with SCD and ensuring access to important and
innovative new medicines.”
GBT is committed to ensuring that people with SCD who are
prescribed Oxbryta have help accessing the medicine. The company has
established GBT Source™, a comprehensive program for patients who are
prescribed Oxbryta that provides a wide range of practical, educational and
financial support customized to each patient’s needs. More information is
available at www.Oxbryta.com
[FDA News Release, November 15, 2019] – FDA has approved Adakveo
(crizanlizumab), previously known as SEG101, to reduce the frequency of
vaso-occlusive crises (VOCs), or pain crises, in adult and pediatric patients
aged 16 years and older with sickle cell disease.
“Adakveo is the first targeted therapy approved for sickle
cell disease, specifically inhibiting selectin, a substance that contributes to
cells sticking together and leads to vaso-occlusive crisis,” said Richard
Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting
director of the Office of Oncologic Diseases in the FDA’s Center for Drug
Evaluation and Research. “Vaso-occlusive crisis can be extremely painful and is
a frequent reason for emergency department visits and hospitalization for
patients with sickle cell disease.”
Sickle cell disease is an inherited blood disorder in which
the red blood cells are abnormally shaped (in a crescent or “sickle”
shape), which restricts the flow in blood vessels and limits oxygen delivery to
the body’s tissues, leading to severe pain and organ damage. It is also
characterized by severe chronic inflammation that results in vaso-occlusive
crisis where patients experience episodes of extreme pain and organ damage. The
disease occurs most often in African-Americans, where 1 out of every 365 babies
born have the disease.
The FDA’s decision to approve Adakveo 5 mg/kg is based on
results of the 52-week, randomized, placebo-controlled SUSTAIN trial, which
showed that Adakveo significantly lowered the median annual rate of VOCs to
1.63 vs 2.98 compared to placebo (P=.010), which is equivalent to a 45%
reduction. Reductions in the frequency of VOCs were observed among patients
regardless of sickle cell disease genotype and/or hydroxyurea use.
“We know this drug can decrease the frequency of sickle cell
pain crises in a significant and clinically meaningful way,” said Kenneth
Ataga, MD, Director, Center for Sickle Cell Disease, University of Tennessee
Health Science Center at Memphis, and Principal Investigator of the SUSTAIN
trial. “The approval of crizanlizumab is an important advancement for people
living with this very difficult condition.”
The most common adverse reactions among
crizanlizumab-treated patients (incidence > 10%) were nausea (18%),
arthralgia (18%), back pain (15%) and pyrexia (11%).
Novartis has priced Adakveo, which is dosed by weight, at a
wholesale acquisition cost (WAC) of $2,357 per vial. Based on dosing
recommendation, most patients would take between 3 and 4 vials per month. This translates
to WACs of $7,000 to $9,500 per month. The medicine is expected to be available
to patients in the coming weeks.