The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
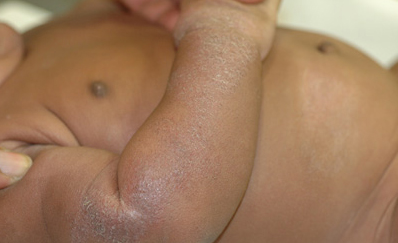
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).